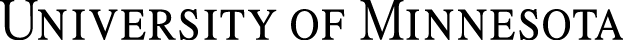
20 Medical Device Product Security
Randy Schiestl; Ken Hoyme; and Bill Aerts
Introduction
Medical device security has always been of paramount importance to the healthcare industry. Today, defining and meeting security requirements is becoming even more essential. Security threats combined with increased connectivity and device capability create new ecosystem inputs and outputs to address. In addition, new stakeholders, including researchers, with genuine patient and healthcare system interests are emerging.
Medical device suppliers, regulators, universities, hospitals and the broader healthcare community are responding with commitment and shared accountability. Both commercialized products and those under development are included in the scope of product security. For all medical device products and services, up-front security planning and implementation throughout the total product life cycle is critical for the protection of patient information, the performance of the devices and the advancement of healthcare solutions. The figure below shows the product life cycle and a few key security related inputs.
Elements of a Product Security Program
Product Security Awareness
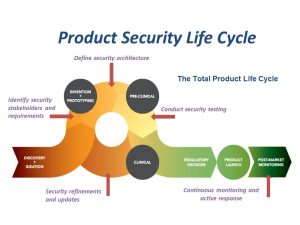
There was a time when medical devices operated independent of networks and were operated only via interaction with doctors and patients. Today, technological advancements have enhanced the life-saving capacity of medical devices with features like wireless connectivity, remote monitoring, and near-field communication. But these advancements can also serve as points of exposure for the devices that are operating in higher risk environments. Every networked system is vulnerable to some degree, so it is logical that the evolution of medical devices in the Internet of Medical Things (IoMT) has created opportunities for attackers to exploit connectivity in the healthcare sector in the clinical setting as well as in home and on personal devices. Making connected devices and the information they use less vulnerable to attack has become a difficult challenge for device manufacturers, Health Delivery Organizations (HDOs) and patients.
The use of connectivity in medical devices has been accelerating. As each device has some level of vulnerability, the threats and risks associated with those devices and systems is increasing as these vulnerabilities get exposed over a network. This situation is exacerbated by the fact that even newer devices that are currently being manufactured and sold still have significant vulnerabilities.
Security vulnerability in a medical device presents an even higher risk than those that we know for traditional internet-based technology. Where key security risks have traditionally been around Confidentially of data, Integrity of function and data and Availability of system and data (the CIA triad), when dealing with healthcare one must also include risks to hospital networks and ultimately patient safety. Any security event that can have potential impact on patient safety must be included in threat and risk management planning for the device that ultimately prescribes required security controls.
The new environment of high connectivity and high vulnerability is driving change in healthcare delivery. Devices that may not protect patient data from disclosure, that may become unexpectedly unavailable, or that cannot protect the accuracy of vital information, may present additional safety risk to patients or threaten the ability of our healthcare systems to deliver timely, reliable and quality healthcare. From healthcare as a business perspective, highly secure medical devices and systems add value for health delivery organizations and health care providers, by reducing time and money spent preventing security events, reducing risks to patients, protecting the efficient functioning of the hospital or clinic providing care, and reducing legal liability due to both data breaches and potential patient harm.
There are many examples that demonstrate this point. A device or system that is susceptible to the latest internet worm or ransomware could mean significant outages for healthcare providers, as well as high cleanup and recovery costs. A device or system that allows its communication or data to be tampered with, can lead to misdiagnosis, incorrect treatment, or costly impact on hospital finances. There have also been documented cases of attackers using a vulnerability in a network connected medical device to launch an attack across the hospital network to acquire valuable patient healthcare records.
There are at least three primary sources of device vulnerabilities. A first is from design decisions made by the manufacturer. For example, if a manufacturer decides not to encrypt patient data on the device, that vulnerability is present in all devices, until and unless the manufacturer performs a design change to add encryption. Identifying design vulnerabilities involves “threat modeling” which is discussed below.
A second vulnerability source is from implementation errors during the development of the device by the manufacturer. A classic example is a “buffer overflow” error, where missing range checks on a buffer allows an attacker to push data beyond the buffer’s limits and gain an entry point. These errors are reduced by coding standards, code reviews, and tools such as static checkers.
A third vulnerability source is from vulnerabilities contained in the third-party software used in the device, such as a Microsoft Windows Operating System. When Microsoft issues patches for a newly identified vulnerability, devices with that OS have that vulnerability until they are patched.
Product Security Stakeholders
The healthcare system is an enormous and complex set of business processes, workflows, systems, providers and technology with many stakeholders engaged to deliver patient care. Not surprisingly, improving medical device security and safety is also complex and impacts many stakeholders, as well as requiring many people and groups to make possible significant reduction of risk.
- Patients have the largest stake in this challenge as vulnerable medical devices can lead to issues with confidentiality, integrity, availability and safety that can directly impact the patient. The scope and potential impact to patients continues to grow as more healthcare technology is used in-body, on-body, or with mobile technology that can often be self-monitored and/or administered by the patient.
- Clinicians that deliver care can be impacted in significant ways and need to play a big role in finding solutions. Obviously, any clinician using a device or system that is unexpectedly unavailable, unreliable, malfunctioning or presents new patient safety risks must not only realize that these kinds of events are possible, but know how to respond to the situation and alter care delivery in a way that protects and improves the patient. Further, this stakeholder must be informed about the security risks associated with a device and follow good security practices in practice to reduce vulnerability and prevent incidents from occurring.
- Health delivery organizations such as hospitals, clinics, emergency and urgent care locations, and lab providers must do everything possible to prevent medical device security incidents and respond quickly when they do occur. The challenge is especially difficult because there is a broad mix of old and new technology in place, the networks are large, and there is a variety of different types of medical devices, systems, sensors, monitors, and other devices that are connected to a network. Complexity is the enemy of good security. HDOs have been working with regulators and device manufacturers to educate them on the environments that the devices must operate in and define specific security requirements for the devices and systems they want to purchase and deploy.
- The United States Food & Drug Administration (FDA) and other global agencies have taken significant steps in regulating medical device security and safety. The most significant US examples are two FDA guidance documents released in 2014 and 2016. The first, Content of Premarket Submissions for Management of Cybersecurity in Medical Devices [1] provides processes to integrate security into the design and development of medical devices. The second, Postmarket Management of Cybersecurity in Medical Devices [2] outlines processes and capabilities that are necessary to manage the on-going security of medical devices once they are in the marketplace. These documents have led to important change and improvement across the industry. Led by the FDA example, regulatory agencies around the world are also building their regulatory requirements for medical device security. In 2020, the International Medical Device Regulators Forum (IMDRF) issued their Principles and Practices for Medical Device Cybersecurity [3] document that is aligned with the FDA guidance documents. The FDA 2014 Premarket guidance document is under review and update at the time of this writing.
- The medical device manufacturer (MDM) is another stakeholder in medical device security. Like all technology from hardware to software to networks, security needs to be built in, not added later. That is especially true for medical devices. Manufacturers are moving to add security planning and design into all their product lifecycles and training as well as creating global security organizations to driving security innovation. This kind of organizational change is generally incorporated over several years. Additionally, manufacturers are getting more pressure to build secure devices not only because of increased regulatory pressure, but because their customers (HDOs and patients) are demanding more proof of security in the products they buy, and better support for security issues that are discovered affecting products in the field.
- Researchers are playing a role in exposing vulnerabilities in medical devices and innovating solutions. Independent researchers have established themselves as a major stakeholder by continuously discovering vulnerabilities and proactively bringing them to the attention of manufacturers and regulatory agencies.
- Academic researchers continue to play an important role in both the medical device security and healthcare front. Significant research on security technologies and solutions have been conducted by colleges and universities in the US and other parts of the world. Universities and colleges are increasingly developing programs and curriculum geared towards educating students who graduate and staff the cybersecurity workforce, helping to close the negative unemployment ratio. Given the current challenges that remain to be solved in the industry, the demand for cybersecurity knowledge and workforce will continue to increase and as such academics will play a critical role in meeting this demand.
Program Management Best Practices
As the security risk to medical devices has continued to expand, manufacturers have come to realize that they need to develop and implement dedicated programs within their companies to deal directly with this challenge and change both the culture, and process of developing and supporting secure technology. In recent years, most of the large and many mid-sized manufacturers have built medical device security programs, but few small device makers have the ability to dedicate the necessary resources. In any case, best practices and support resources are emerging as more programs continue to be built.
For a program to succeed, clear and frequent support from the executive and senior leadership at the company is essential. That includes regular and direct communication on the importance, a long-term commitment of financial and other resources, and demanding accountability. Governance practices and controls must also be in place including policies, metrics, regular reporting and independent reviews. To have the desired impact, the program must take a cross functional, and company-wide approach. Virtually all major functional groups in the company have a role to play.
The Healthcare & Public Health Sector Coordinating Council’s Joint Cybersecurity Working Group has published a free Joint Security Plan (JSP) [4], which presents a framework and recommended implementation steps geared towards small manufacturers so they can easily establish a security program as an integral component of their quality system.
Medical device engineers and software developers are highly talented and dedicated people. They have been trained and immersed in technologies, practices and standards designed to ensure that patient safety is the number one priority in all their work. A substantial amount of effort and resources should be dedicated to raising awareness and providing training to product developers that increase their understanding of the real security risks that exist, introduce new risk management approaches to build security into the product development process, and develop new technologies, techniques and controls that can deliver the highest levels of protection.
Because of the unique nature of security in devices, different risk management methodologies for use in product development and ongoing support are warranted. While there are many additional and useful standards and methodologies used for management of safety risk, ANSI/AAAMI/ISO 14971:2007 Medical Devices – Application of Risk Management to Medical Devices [5] is highly regarded by the FDA and widely used methodology for manufacturers to use in managing safety risk. In 2016, AAMI released an associated Technical Information Report titled AAMI TIR57: Principles for medical device security – Risk Management [6]. TIR57 blends security and safety risk management by showing how to apply the principles in 14971 to security threats that may impact the confidentiality, integrity, and the availability of a medical device or the information processed by the device.

Another organizational practice that is important to include in a program is to develop strong partnership and process integration between the Information Technology (IT), Device Engineering (R&D) and Quality groups. As healthcare becomes more connected, most medical device products/systems now have more standard IT components, each with its own security concerns. Examples include Bluetooth, Windows or other familiar operating systems, standard software components, network protocols, data stored in the cloud and mobile devices. These are all technologies that IT groups have familiarity and have implemented many times in their business systems and enterprise infrastructure. While product engineers know everything about their device, some of these technologies may be new to them, or the R&D group is not equipped to implement large connected systems. If these two organizations are working together, each brings their specific expertise and capabilities, which in the end will deliver better, more secure products.
As security researchers, the manufacturers themselves and other groups discover potential security vulnerabilities in existing products, manufacturers need to develop a coordinated disclosure policy and process. The goal is to ensure that when these potential security vulnerabilities are brought forward, there is a fair, consistent, and reliable process to handle this type of discovery quickly and appropriately. Additional information on this topic can be found in the FDA post-market guidance.
Finally, no one manufacturer is going to solve all their security issues on their own. Fortunately, there is a large community of stakeholders and groups that exist for manufacturers to engage with. Examples include professional organizations, standards bodies, regulators, the Health Information Sharing & Analysis Center (H-ISAC) [7], the media, public conferences and educational offerings. Reaching out and getting engaged with these groups and the people in them helps every manufacturer advance security, as well as improving the image of the company, as it will be viewed as a visible contributor in advancing our ability to secure medical devices.
FDA Pre-Market and Post-Market Guidance
In 2012, the Government Accounting Office (GAO), responding to reports from security researchers about potential vulnerabilities in medical devices, issued a report [8] directing the FDA to improve their oversight of the pre-market analysis of security risk and in the post-market monitoring and device patching processes . In 2014 the FDA released their final guidance on the Content of Premarket Submissions for Management of Cybersecurity in Medical Devices (FDA, 2014) and in 2016, final guidance on the Postmarket Management of Cybersecurity in Medical Devices (FDA, 2016). While FDA guidance documents are nonbinding, security representatives from the agency have made it clear that they consider these guidances as a way to meet the Quality System Regulations (QSR). While a manufacturer may offer an alternate method to meet the information in the guidance, to ignore them is to risk significant delay in device approval and may lead to future audit risks for the post-market security risk management plan.
The pre-market document is applicable to broad set of submissions for devices that contain software, firmware or programmable logic as well as software that is a medical device (in-scope devices). The post-market guidance extends the list of in-scope devices to include mobile medical applications, devices that are part of an interoperable system, and to legacy devices – those that are already on the market or in use.
The pre-market guidance directs the manufacturer to conduct a thorough security risk assessment to identify risks that could lead to patient harm and mitigate those risks to an acceptable level. It suggests several classes of security controls to consider, organized along NIST’s Cyber Security Framework (CSF) [9]. Finally, it identifies the types of cybersecurity documentation that the agency expects to be in a pre-market submission.
The post-market guidance is more complex. In this case, the agency is working to define when a manufacturer can patch a security issue quickly without pre-approval, and when the normal processes must be followed. They introduce the concept of routine updates and patches to represent updates only for security where patient harm has not occurred. They introduce the concepts of controlled and uncontrolled risk, where uncontrolled is when there is unacceptable residual risk of patient harm. Finally, the concept of compensating controls is used to represent things the end user can do to reduce risk while a patch is being prepared and verified; for example, the temporary removal of the device from a hospital network, if the attack vector is network-based.
This guidance document also directs manufacturers to monitor various sources for information about new vulnerabilities that may impact a device, for example, if the device uses a commercial operating system such as Windows. The guidance also recommends consideration of methods for monitoring devices for detecting the presence of vulnerability. Further, they recommend manufacturers establish a coordinated vulnerability disclosure process to receive input from researchers and other parties that may believe they have discovered a vulnerability in one of your devices.
Finally, the guidance document allows manufacturers to publish compensating controls within 30 days and issue a fix within 60 days. Along with other criteria being met, the procedure is for controlled vulnerabilities. 21 CFR 806, Medical Device Correction and Removals is not needed and can be replaced with annual reporting. These guidance documents will continue to evolve, so manufacturers should consult the FDA website for current copies if you are developing an in-scope medical device.
In 2019, AAMI released an associated Technical Information Report titled AAMI TIR97: Principles for medical device security – Postmarket risk management for device manufacturers [10]. TIR97 details the various security activities that a manufacturer should plan to implement to support medical devices that have been deployed to the field.

Security by Design
Quality System
Aspects of the previously discussed FDA guidance documents should be incorporated into the policies, procedures and work instructions in the manufacturer’s quality system, such as:
- Requirements for classes of devices to be developed should include security.
- The security risk management expectations should be incorporated into the quality system, with appropriate links or mapping to the safety risk management activities.
- Security (penetration) testing should be considered an extension of the process for requirements-based testing – as a capstone activity
- Postmarket monitoring Cybersecurity signal (security issues) investigation processes should be incorporated into the postmarket support activities for medical devices.
While safety and security are related, they are not the same. There are traditional safety risks that have no relationship to security; for example, hardware component failure. There are security risks that have no impact on safety; for example, a data breach does not fit the definition of harm from ISO 14971. But there are risks that can interact (for example ransomware can make a device unavailable) and if it was delivering therapy to a patient (for example, an infusion pump) that therapy will be impacted.
Product Security Requirements
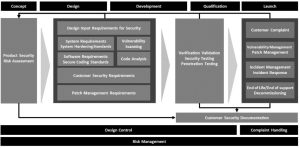
During medical device systems development, the typical starting point is to create design input requirements that represent what the device should do from the view of the end user. In the case of a software driven or connected medical device, the designer should recognize that one of these end-user stakeholders is the one responsible for securing the device according to the policies of that organization. So, even at the design input requirements level, requirements for confidentiality, integrity, availability, authentication and authorization must be considered. The security categories contained in IEC/TR 80001-2-2: Application of risk management for IT-networks incorporating medical devices – Part 2-2: Guidance for the communication of medical device security needs, risks and controls [11] offer guidance for what to consider during the early stages of device development.
System design requirements can contain additional detail, including specifics of encryption, key sizes, protocol specifics, certificate standards, and security architecture requirements for issues such as key storage, secure code updating, and security logging details. Requirements must be testable, which means in terms of shalls (what the system will do), and not in terms of shall nots (what the system will never do.) Shall nots are typically untestable. So instead of, “the system shall not let an attacker perform a brute force log in attack,” use “the system shall lock an account out for 15 minutes if 10 consecutive failed login attempts are made against a single account.”
Security Risk Management
Central to development of a secure medical device is the implementation of a robust security risk management process. It should start at the concept phase when first considering the product specifications and performing marketing studies. During early phase development, the security risk assessment drives the creation of the security requirements, both at the design input and system design levels. System Engineering is critical to implement a comprehensive approach to product security. Typically, system engineers within R&D, where product design ownership often resides, drive the broad product performance requirements as well as security architecture and controls.
The security risk management process should be comprehensive, in terms of security impacts. It should not be limited to security risks that just impact patient safety because business risks, such as financial penalties for data breaches, and reputational damage should be considered. The risk assessment process should also consider risks where a device could put other systems on the connected network at risk. For example, malware that targets health records might use a weak medical device to pivot and attack other systems on the network as they seek the records stored elsewhere in the system.
Security risk management should also include a threat modeling exercise, where the overall systems architecture is mapped out and the location of key assets identified. Understanding the layers of defense against threats that may be external or internal help the analyst understand what mitigating controls are needed to protect critical assets. Understand that in a medical device, where safety is of paramount concern, knowing how to maintain the integrity and availability of key assets and device essential performance must be a primary concern.
During development, the device design team should seek to balance safety, security and usability. For example, the need for emergency access might limit the requirement to authenticate for a normal user account while authentication may still be required for administrative accounts. Usability concerns may drive one to synchronize with hospital ID management servers (for example, Active Directory or LDAP) so that users are not required to remember different passwords and more streamlined user account management and auditing.
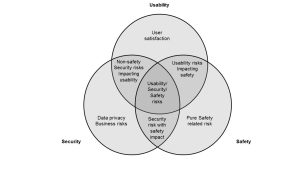
Developers should be aware of ISO 14971:2010 , which is the overarching standard for safety risk management. AAMI TIR57:2016 uses the 14971 framework to teach how to conduct security risk management drawing on NIST SP800-30 Rev 1: Guide for Conducting Risk Assessments [12]. The usability analysis of medical devices is driven by IEC 62366-1: Medical devices – Part 1: Application of usability engineering to medical devices [13].
The risk analyses should be updated during product development as design decisions are made that might impact the results. It is also the basis for evaluating risk during post-market, in order to determine if a change impacts the previous risk assessment.
Threat Modeling
The exercise of threat modeling (also known as security architecture analysis) is becoming recognized by regulators such as the FDA, as a “Best Practice” for ensuring that the security risk analysis is thorough. Threat modeling describes a wide range of methodologies meant to understand the security architecture of a device and how an attacker might penetrate its defenses and impact confidentiality, integrity or availability. The STRIDE methodology promoted by Microsoft is one of the easier ones to adopt for a new organization. Microsoft also provides a free Threat Modeling Tool based on this methodology [14].
The Medical Device Innovation Consortium (MDIC) has been funded by the FDA to develop a threat modeling bootcamp, and in conjunction with MITRE, a threat modeling playbook, which should be available in late 2021 [15].
Secure System Software
There are additional steps that should be taken to ensure that the custom software developed to implement the medical device application is secure. The first step is to establish secure coding standards. The SEI CERT has several examples for popular programming languages [16].
Being aware of the most frequent errors to avoid is also helpful for the team. OWASP has their annual Top 10 [17], SANS has their Top 25 [18]. Other sources can be found, including the Common Weakness Enumeration (CWE) list [19] that describes the typical ways that security weaknesses can be unintentionally coded into a system.
Commercial and open source static checking tools are available that specifically look for such coding errors in custom software. Introducing such tools into the development process and resolving the errors that are noted by these tools is becoming a best practice in this industry.
Security Testing
Typical medical device testing is requirements-based, to ensure that the device does all of the things the requirements say it should do. But understanding whether there are exploitable vulnerabilities that could interfere with the device delivering its intended behavior requires a different form of testing. Two testing classes are typically performed: robustness testing and penetration testing.
Robustness testing tries to determine if the device has been designed defensively. Typically, the command and response stream that flows over interfaces is designed according to a specific protocol. Protocols usually establish rules on the acceptable values in certain fields, lengths of buffers, etc. One form of robustness testing, called fuzz testing, attempts to break those rules and send malformed messages to the device to see how it responds. A defensively programmed device rejects those commands and continues to operate normally, but others may exhibit other behaviors, including a crash. Ensuring a device does not unreasonably trust the system it is interacting with to always play by the rules is a basic precept of secure device design.
For penetration testing, a security expert takes the role of a hacker and using all the tools and skills at a hacker’s disposal, applies them to a device to see if they can penetrate its functionality. While large organizations may have this skill internally, most companies will contract this task out to skilled third parties. Many large hospitals now expect manufacturers to do this kind of testing in a regular cadence (because of evolving threats) and want to review the reports for what was found.
Post-Market Product Security
As previously referenced, refer to the 2016 FDA’s post-market guidance, AAMI’s TIR97 and HSCC JCWG’s Joint Security Plan for additional details on post-market product security management.
Sources of Cybersecurity Signals
Typically, device manufacturers get signals about the performance of their devices in the field from two primary sources: customer complaints and returned product. As the connected world has evolved, complaints may come from sources such as social media, blogs, and news articles; not just calls into a complaint line. But it is still discrete to a single device.
With connected devices, signals that might be indicative of a cybersecurity issue can come from additional sources such as security researchers who specifically analyze a copy of a device; vulnerability announcements from developers of third party software used in the device; periodic post-market penetration testing (for example, it is considered best practice to repeat penetration testing as hacker skills keep changing); and log information from devices in the field (for example, another best practice is to put security logging in a device and define a path for that information to get back to the manufacturer.) Manufacturers of devices that are connected cannot simply rely on the old mechanisms to learn of vulnerabilities. Processes must be put in place to respond to these new signal sources.
Coordinated Vulnerability Disclosure
When a researcher or other external party believes they have identified a specific vulnerability that could be exploited to make a device malfunction, they will usually seek a path to communicate that to the manufacturer. Experience has shown this to be a difficult process, as manufacturers do not like being told their devices are not perfect and worry about the motivations of those who reach out. Experience by those who have been open to working with these external entities have found them to generally want the devices to improve and usually are happy to know they have had a positive impact on patient care.
In the recent post-market guidance, the FDA has indicated their expectations that manufacturers implement a “Coordinated Vulnerability Disclosure” process. They recognize ISO/IEC 29147: Information technology – Security techniques – Vulnerability disclosure [20] as a standard method of doing this. Basically, it ensures that manufacturers receive vulnerability inputs and evaluate them for confirmation and risk impact. Based on that impact they determine a plan for patching, and coordinate with the researcher on that plan. The point of “coordinated” is that any communication to the outside world comes from the company, the researcher and any government entities at the same time, and usually when a patch for the issue is available.
The goal of coordinated disclosure is to ensure that the fix is available before the vulnerability is known broadly . Be aware that there are limits in how long the time from disclosure to the time to fix is tolerated. If manufacturers are perceived as trying to bury an issue using the disclosure process, the researcher may go public without a fix being ready. It is best to be transparent with the researcher and with the government agencies that may be involved, and to negotiate a reasonable response timeline.
Vulnerability Monitoring
Separate from learning about vulnerabilities in the device’s custom code, if it depends on third party code, for example, operating system, libraries or frameworks, it is likely that vulnerabilities in those software components are identified and fixed by their manufacturer independent of any release cycle for the medical device that depends on them.
If the device manufacturer explicitly contracts for third party software, contracting language should define the obligation for the supplier to communicate on newly discovered vulnerabilities and establish expectations on the timing to produce updates.
Larger third-party sources communicate their vulnerabilities and fixes through national databases or other internal mechanisms. Manufacturers should establish a means to regularly monitor these sources to learn about new vulnerabilities that might impact fielded devices and perform risk assessment to determine if a patch is needed, and with what urgency.
There are various commercial tools that can automate this monitoring function. The third-party dependencies are discovered for a device, and then the databases are monitored for new vulnerabilities in the software contained in that device.
Patching
Traditionally, a software-based device was released without a robust plan to patch unless some unexpected safety issue arose. With the thoroughness of requirements-based testing, that was infrequent.
With the increasing interconnectivity of devices that incorporate vulnerable third-party code targeted by common malware, there should be an expectation of a constant patch cadence. If that is done, when vulnerabilities are identified, they can be assessed whether an emergency push needs to be done to resolve, or if it can simply be scheduled for the next planned update. Without a periodic update plan, all patches are off-line emergencies.
Patches need to be distributed securely. Establishing a patch mechanism can be a vulnerability on its own if the patches are not authenticated as coming from the manufacturer. In this environment, authentication needs to be cryptographic.
Secure Disposal
When it is time to take a device out of commission, the manufacturer should have designed provisions to ensure that any protected information in the device can be securely erased. There is a business risk to the manufacturer and the end user if such information ends up in a device being sold on an on-line auction site.
This is also useful if a device is being transferred from one facility to another (for example, a device that is leased). Hospitals will want to know their data has been securely removed before returning it for its next use.
Summary
With increased complexity and increased threats, product security has become even more important to product design and product sustainability. System engineers must capture and implement robust security and safety requirements. All connected devices have known or unknown vulnerabilities that existed at product launch, and additional vulnerabilities that are created throughout their life cycle. All stakeholders in the medical device ecosystem share the responsibility to ensure security and safety for patients world-wide.
Challenges and Areas of Future Opportunity
HDO Network Connectivity
There is immense potential for IoMT to increase accessibility solutions and analytics capability in the healthcare sector, however, access to the HDO network and/or Internet remains a constrain due to stringent network security requirements. Manufacturers who are best positioned to address the requirements need to know about these requirements in advance so that they can bake them in the device’s design early in the development phase. Unfortunately, manufacturers have limited visibility into hospitals Network and cybersecurity policies and other requirements. In cases where they get this input early enough to design them in, there’s often a very high likelihood that the requirements would be stale by the time the product goes to market. HDO’s are constantly having to revise their requirements to keep up with the changing threat landscape and increased attacks against their networks and data.
Standardization of network requirements across HDOs would go a long way in easing some of these challenges, however, this is hard to achieve due to the disparate use cases coupled with varying technologies in IT infrastructure across hospital systems. For example, some hospitals may use 802.1X standard that requires certificates to authenticate to LAN or WLAN, while others may not. Ultimately, as both stakeholders navigate connectivity while balancing risk, innovation and potential efficiencies get stymied.
HDO Security Questionnaires and Agreements
Transparency and exchange of cybersecurity information on device security is another area that still needs work, granted there has been great progress made in recent years. More HDOs, especially larger HDOs and Group Purchasing Organizations (GPOs) now require MDMs to agree to legally binding cybersecurity contracts before purchasing their devices. The language in these contracts is tied to their respective hospital security requirements or industry best practices, geared towards reducing the amount of risk and transferring liability in case of data breaches or network compromise as a result of lax security on devices from a manufacturer. These contractual requirements lead to delays in getting medical devices on the hands of doctors in a timely manner, creating frustrations on both ends as everyone tries to find a compromise often through back and forth redlines.
Additionally, there is an upward trend in the number of questionnaire forms send by HDOs to MDMs during purchasing, an indication of increased need for device security information needed by hospital IT in order to assess for potential risk. The questionnaires are on security controls and architecture of the medical devices and can be as lengthy as two to three hundred questions, with some requesting supporting artifacts such as network diagrams, certificates or whitepapers to be attached to the responses. Every request, while similar in nature, require the information to be providing in disparate formats – online portals, Excel spreadsheets, PDF, Word documents etc. This often takes a while for MDMs to complete and return, as it requires collaboration from multiple internal teams such as legal, R&D, corporate IT, etc. to provide or review responses in their respective areas.
NEMA/MITA led the development of the Manufacturer Disclosure Statement for Medical Device Security (MDS2) [21], a voluntary standard that provides information for security risk management within HDOs. The goal was to provide standardized information on security control features integrated within medical devices and has been revised several times, each time with additional questions. The latest version released in 2019 has over one hundred and sixty-five questions. Unfortunately, as a result of constantly evolving cyber threats, the rate and expansiveness of questions and information the HDOs seek from MDMs, particularly for network connected, continues to increase. More transparency and collaboration will be needed to tackle the fundamental problem of managing risk, and the exchange of cybersecurity information.
Security Requirements
MDMs continue to struggle to find a standardized set of common security requirements that fulfil most of HDO cybersecurity compliance requirements. The challenge is partly due to the vast categories and use cases of medical device technologies with varying capabilities and limitations, which tend to vary depending on the size of a hospital system or the maturity of their IT infrastructure. Some of the areas that could use standardization include authentication, logging, incident response (between HDO and MDM), etc. to name just a few. To address this need, additional collaboration between all stakeholders will be needed to come up with solutions through dedicated workgroups and forums
While these are representative examples of areas of opportunity, it is expected that they will keep evolving with new ones arising as the threat landscape, technologies and user needs continue to evolve. The good news is that healthcare forums and member-based organizations have been formed, such as the H-ISAC, MDIC, the Archimedes Center for Healthcare and Device Security [22], and others that are helping to bring stakeholders together to collaborate on common solutions to tackle existing and emerging challenges such as the ones noted above.
The global product cybersecurity community continues to collaborate and innovate to identify approaches and solutions that mitigate cyber risk. Universities, governments, consortia and service companies are beginning to develop courses and curricula that focus on infrastructure and product-specific cybersecurity education and research. Additional trained resources are needed in industry and in government to meet the growing demand of medical device manufacturers and healthcare organizations. The field of cybersecurity continues to evolve and mature as we share information and best practices to serve patients, physicians and clinicians worldwide.
- FDA, “Content of premarket submissions for management of cybersecurity in medical devices,” Food & Drug Administration, Tech. Rep. FDA-2013-D-0616, 2014. [Online]. Available: www.fda.gov/media/86174/download. ↵
- FDA, “Postmarket management of cybersecurity in medical devices, ”Food & Drug Administration, Tech. Rep. FDA-2015-D-5105, 2016. [Online]. Available: www.fda.gov/media/95862/download. ↵
- IMDRF, “Principles and practices for medical device cybersecurity,” International Medical Device Regulators Forum, Tech. Rep. IMDRF/CYBER WG/N60FINAL, 2020. [Online]. Available: www.imdrf.org/docs/ imdrf/final/technical/imdrf-tech-200318-pp-mdcn60.pdf. ↵
- Health Sector Coordinating Council - Joint Cybersecurity Working Group, Joint security plan (JSP). [Online]. Available: healthsectorcouncil.org/the-joint-security-plan/.76 ↵
- ANSI/AAMI/ISO 14971, Medical devices - application of risk management to medical devices, 2010. ↵
- AAMI/TIR57, Principles for medical device security - risk management, 2016. ↵
- Health information sharing & analysis center. [Online]. Available: hisac.org/. ↵
- General Accounting Office, Medical devices: FDA should expand its consideration of information security for certain types of devices, 2012.[Online]. Available: gao.gov/assets/650/647767.pdf. ↵
- NIST, “Cybersecurity framework,” Nation Institute of Standards and Technology, Tech. Rep. NIST-CSF-v1.1, 2018. [Online]. Available: nvlpubs.nist.gov/nistpubs/CSWP/NIST.CSWP.04162018.pdf. ↵
- AAMI/TIR97, Principles for medical device security - postmarket risk management for device manufacturers, 2019. ↵
- IEC TR 80001-2-2, Application of risk management for IT-networks incorporating medical devices - part 2-2: Guidance for the disclosure and communication of medical device security needs, risks and controls, 2012. ↵
- NIST, SP 800-30 Rev. 1, Guide for conducting risk assessments, 2012. ↵
- IEC 62366-1, Medical devices - part 1: Application of usability engineering to medical devices, 2015. ↵
- Microsoft threat modeling tool 2016. [Online]. Available: www.microsoft.com/en-us/download/details.aspx?id=49168. ↵
- MDIC, Medical device innovation consortium cybersecurity initiatives.[Online]. Available: mdic.org/program/cybersecurity/. ↵
- Software Engineering Institute, SEI CERT coding standards, 2020. [Online]. Available: wiki.sei.cmu.edu/confluence/display/seccode/SEI+CERT+Coding+Standards. ↵
- Open Web Application Security Project, OWASP top ten, 2017. [Online]. Available: owasp.org/www-project-top-ten/. ↵
- SANS Institute, CWE/SANS top 25 most dangerous software errors,2011. [Online]. Available: www.sans.org/top25-softwareerrors/. ↵
- MITRE, Common weakness enumeration (CWE), 2020. [Online]. Available: cwe.mitre.org/. ↵
- ISO/IEC 29147, Information technology — security techniques — vulnerability disclosure, 2018.References 77 ↵
- N. E. M. Association, Manufacturer disclosure statement for medical device security (mds2), 2020. [Online]. Available: www.nema.org/Standards/view/Manufacturer-Disclosure-Statementfor-Medical-Device-Security. ↵
- Archimedes center for healthcare and device security. [Online]. Available:www.secure-medicine.org. ↵
