18 Micra™ Development and Regulatory Process: A Case Study
David Brooke and Michael Eggen
Introduction
A traditional single-chamber pacing system consists of a pacemaker, sometimes called a can, implanted in the subcutaneous pectoral pocket that is electrically connected to a transvenous pacing lead placed in the right ventricle. Although such a system has provided pacing therapy for almost 60 years, the subcutaneous pocket and leads can be a source for complications such as lead dislodgement, lead fracture, lead or pocket infection, pocket hematoma, and vascular obstructions. To reduce or eliminate these complications, Medtronic developed the first FDA approved leadless pacing system. The Micra™ Transcatheter Pacing System (TPS)12 is a single chamber, leadless pacemaker implanted entirely within the right ventricle of the heart, eliminating the need for a device pocket and insertion of a pacing lead. The 0.8 cc device is implanted percutaneously via the femoral vein using a 23 French introducer and delivery system. As such, the device is affixed to the heart via four nitinol fixation tines, which provide device fixation and a stable electrode-myocardial interface for efficient pacing capture. This chapter summarizes the development and regulatory process/timeline that was necessary to bring this pacing technology to the medical device market.

The Micra™ TPS regulatory process began shortly after the development program showed promise as an opportunity to provide pacemaker therapy without the need for pacing leads. Early meetings with global regulatory authorities served to educate regulators, identify potential areas of concern and define clinical study requirements. In today’s global environment there can be significant advantage in conducting clinical studies in multiple geographies simultaneously. Many countries have developed requirements for in-country clinical data in support of market approval so making use of a single global trial rather than multiple in-country trials can be more efficient. Global studies also provide for experience with a variety of implanters, cultures and medical practices. The Micra™ project team decided early in the development process that a global clinical study would be the best approach since it was determined that the miniaturized pacemaker could benefit patients around the world.
The FDA has traditionally driven study design requirements from their rigorous review process focused on safety and effectiveness outcomes. For many years clinical evidence generated for US market approval has been sufficient to support other global geographies. In today’s evolving global regulatory environment, however, Europe and some Asian countries have also begun to establish significant clinical requirements for market approval of new medical devices and therapies that sometimes are not met with traditional US based studies. Other considerations for clinical study designs include the need to provide supporting data for health care economics claims and Medicare coverage. These studies often require comparison to standard of care therapies, which can add significant expense to the clinical trial depending on the device or therapy being developed. It is critical for a company to understand the conditions for reimbursement so that after finally receiving regulatory approval there is assurance that the new technology will be covered by the Centers for Medicare and Medicaid Services (CMS). CMS is the organization that provides oversight and guidance to coverage decisions for the majority of healthcare services in the US. Patients are rarely able to pay out-of-pocket for medical expenses, particularly when new and innovative therapies are involved. Therefore a successful CMS coverage strategy for Micra™ was vital for releasing a sustainable product to the market.
Study Design
The Micra™ transcatheter pacing study was a prospective, nonrandomized, single-study-group, multisite, international clinical study to evaluate the safety and efficacy of the Micra™ Transcatheter Pacing System. 345 Patients were evaluated for adverse events and device function at hospital discharge and at 1, 3, and 6 months and every 6 months thereafter. The study included options for multiple endpoints including a safety and efficacy analysis after 60 patients were followed for 90 days that was used for market approval for European Union and other countries accepting the CE mark. The study’s long term objectives included two primary end points that were assessed at 6 months of follow up. The primary safety end point was freedom from system-related or procedure-related major complications, where the performance benchmark was based on a historical control. The primary efficacy end point was the combination of a low (≤2 V @ 0.24 ms pulse width) and stable (increase of ≤1.5 V from the time of implantation) pacing threshold at the 6-month visit. It was estimated that a sample size of 720 successfully implanted subjects would provide 90% power to test the study’s two primary end-points. The study was considered successful at the first analysis if both primary objectives were met. There was also a long-term safety objective that evaluated freedom from major complications related to the Micra™ system or procedure at 12 months. The first patient in the trial was implanted in December, 2013.
Concurrent with the Micra™ project initiation, the FDA began multifaceted programs to foster innovation with industry, with the goal of finding ways to improve access to new technologies for US patients. One of these programs allowed for a reduced set of clinical data to be submitted in support of market approval. In this provision, manufacturers were allowed to submit an early analysis for approximately half of the total required study cohort and then submit the remainder of the data as a post approval requirement. As a result, by allowing the early clinical data to be used in support of the PMA the Micra™ TPS approval cycle was reduced by nearly a year.
US Regulatory Submission Process
A premarket approval (PMA) submission is required to obtain approval to market new class III devices in the US. A PMA submissions is extensive, including multiple volumes of preclinical testing data, comprehensive risk analyses, animal study data, device labeling and manuals, and human clinical data. Micra™ TPS, like other pacing devices such as intra-cardiac leads, is regulated as a combination product because it contains a steroid drug component that helps reduce scar tissue formation at the tissue contact site. A combination product as defined in 21 CFR 3.2(e), is a product comprised of any combination of a drug and a device; a biological product and a device; a drug and a biological product; or a drug, device, and a biological products.6 Combination devices often require that much more supporting data be included with FDA submissions than non-combination devices.
When large clinical trials are involved such as that required for the Micra™ TPS, the FDA allows a provision for modular submissions. Modular submissions allow the manufacturer to submit much of the required supporting preclinical data while the clinical study is ongoing. Once the final clinical report is issued, it can be submitted and reviewed efficiently along with proposed instruction manuals for the new device. The statutory review cycle is 180 days from receipt of the final PMA submission module. If any significant deficiencies are noted by the agency, however, the FDA review clock is stopped until the manufacturer has resolved the deficiencies. If only minor issues are found that can be reviewed concurrent with the main file the agency can use a “proceed interactively” process where the clock is not stopped. This is determined at the halfway point of the review cycle and the Micra™ TPS was able to benefit from this review option. An overview of the regulatory review process associated with the Micra™ TPS is shown in the figure below.
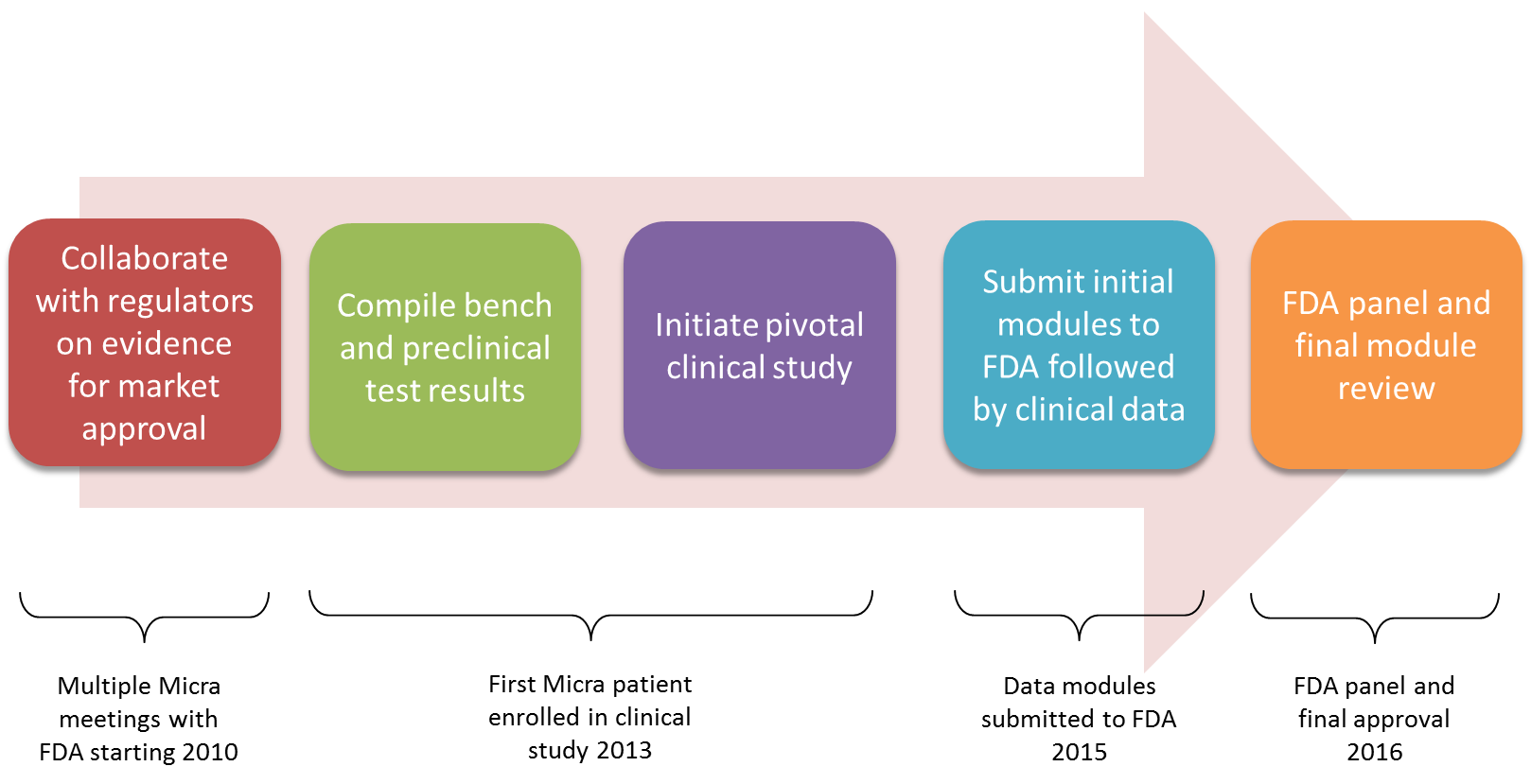
The Micra™ TPS modular submission and related correspondence contained over 56 hard-copy paper volumes with 400-500 pages per volume for a total of over 22,000 total pages. (The FDA began an eCopy program in 2013 which is intended to incrementally reduce the need for paper copies for future submissions.7)
For many new devices and therapies, the FDA initiates an advisory committee review. This is conducted by a panel of independent experts tasked to provide an evaluation of the clinical data and any other supporting data deemed important by the agency. Panel experts include practicing clinicians and biostatisticians in addition to appointed FDA personnel as well as non-voting patient and industry representatives. The panel is typically tasked with determining whether the supporting data is sufficient to conclude that the risks of the proposed new technology are outweighed by the benefits as determined primarily from the clinical results. The panel completes their process by voting at the conclusion of the session and providing this information to the FDA for consideration. The FDA is not obligated to follow the recommendations of the panel, but historically is in agreement.
Companies take these advisory committee meetings seriously. They are important to the approval process and the proceedings are held in a public forum followed immediately by a press release summarizing the results. It is not uncommon for companies to dedicate a great deal of time and resources in preparation and often hire consulting firms specializing in panel proceedings. Companies typically send large delegations including independent clinical and technology experts, executive management representatives, support staff, and personnel preparing for future panel meetings. Panel committees have recurring themes for questions and concerns that evolve as technology, political climate and committee members change over time. It is important to monitor the progression of these changes in preparation for future device approvals.
Micra™ TPS presented a special case with regard to the use of the advisory committee. Since bradycardia pacing therapy was not new and the clinical trial met all endpoints by wide margins, a general issues modified form of panel meeting was conducted. A general issues panel is tasked to answer specific questions from the FDA as deemed necessary and does not vote on risk vs. benefit as is done for a typical PMA focused panel. For leadless pacing technology, the general issues panel was tasked to provide recommendations that would apply to all current and future device manufacturers such as post market follow up clinical criteria, training program requirements and end of life considerations. The panel was not asked to assess risk vs. benefit since the agency was able to provide this assessment within their internal review teams. The general issues panel was supportive of leadless pacing and provided helpful guidance to the FDA in establishing ongoing criteria for adoption of the technology.8 This included guidance to monitor end-of-life experiences in post approval studies since these devices have an expected service life of over 10 years. Pre-market clinical trials are not generally designed to monitor patients for this duration and therefore are evaluated in a post-approval setting until evidence of an adequate safety profile is achieved. This process commonly allows for timely release of novel and innovative therapies to patients while also controlling and monitoring the technology until fully established in the marketplace.
Because of the reduced scale of the general issues panel as applied to the Micra™ TPS regulatory process, the preparation and delegation activities were reduced accordingly. However, many months of preparation were required to adequately represent the supporting data and recommendations for the Micra™ TPS and a large delegation of experts and support staff were in attendance.
Micra™ Clinical Trial Study Results
Enrollment was completed in May 2015, with 744 patients enrolled at 56 centers in 19 countries in North America, Europe, Asia, Australia, and Africa.9 A total of 725 patients underwent an implantation attempt, and 719 were successfully implanted with a device. The study met its primary objectives at the 3 and 6-month time points. The pacing thresholds were low and stable and the patients with transcatheter pacemakers had significantly fewer major complications than did the historical control patients. To support European approval, an early analysis was performed when 60 patients had reached the three-month time point and the initial primary analysis was performed when approximately 300 patients had reached the six-month time point. Because of the program created by the FDA, the initial primary analysis could be used to support the PMA submission.
Micra™ TPS received European CE mark approval in April of 2015, FDA approval in April of 2016 and US CMS approval for reimbursement in 2017. The total development cycle was approximately 8 years. In addition to the extensive work done by the design, development and clinical teams, a large part of the program’s success was due to the close interaction with regulators throughout the development process.
Acknowledgements
The authors acknowledge Verla Laager and Kurt Stromberg for their leadership in the clinical study design and regulatory strategy.
- Micra™ is a trademark of Medtronic PLC.↩︎
- Images reproduced with the permission of Medtronic PLC.↩︎
- Reynolds D, Duray GZ, Omar R, Soejima K, Neuzil P, Zhang S, et al. A Leadless Intracardiac Transcatheter Pacing System. N Engl J Med, 2016 Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643. Epub 2015 Nov 9.↩︎
- https://clinicaltrials.gov/ct2/show/NCT02004873. Accessed October 28th, 2017.↩︎
- Ritter P, Duray GZ, Zhang S, Narasimhan C, Soejima K, Omar R. The rationale and design of the Micra Transcatheter Pacing Study: safety and efficacy of a novel miniaturized pacemaker. Europace, 2015 May;17(5):807-13. doi: 10.1093/europace/euv026. Epub Apr 7, 2015.↩︎
- FDA, Combination Product Definition, available https://tinyurl.com/y7wmsx4a. Accessed October, 2017↩︎
- FDA, eCopy Program for Medical Device Submissions, available https://tinyurl.com/mz2hl2w, Accessed November, 2017.↩︎
- FDA Executive Summary Memorandum, General Issues: Leadless Pacemaker Devices, Feb. 2016, available www.fda.gov, Accessed November, 2017.↩︎
- Reynolds D, Duray GZ, Omar R, Soejima K, Neuzil P, Zhang S, et al. A Leadless Intracardiac Transcatheter Pacing System. N Engl J Med, 2016 Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643. Epub 2015 Nov 9.↩︎
