110 Aging
By the end of this section, you will be able to:
- Explain the physiological explanations for aging.
- Explain the evolutionary explanations for aging.
- Describe how trade-offs between survival and reproduction can result in the evolution of aging.
- Explain the evolution of different life-history strategies.
What is Aging?
Aging is the progressive loss of physiological functions that increases the probability of death. Table 1 gives some data about loss of structure and function in aging.
| Weight of brain | 56% |
| Blood supply to brain | 80 |
| Output of heart at rest | 70 |
| Number of glomeruli in kidney | 56 |
| Glomerular filtration rate | 69 |
| Speed of return to normal pH of blood after displacement | 17 |
| Number of taste buds | 36 |
| Vital capacity | 56 |
| Strength of hand grip | 55 |
| Maximum O2 uptake during exercise | 40 |
| Number of axons in spinal nerve | 63 |
| Velocity of nerve impulse | 90 |
| Body weight | 88 |
The decline in function certainly occurs within cells. This is especially true of cells that are no longer in the cell cycle:
- neurons in the brain
- skeletal and cardiac muscle
- kidney cells
Tissue and organs made of cells that are replenished by mitosis throughout life. Blood and intestinal epithelium show far fewer signs of aging.
In the natural world, very few animals live long enough to show signs of aging. Random mortality from starvation, predation, infectious disease and a harsh environment (e.g., cold) kills off most animals long before they begin to show signs of aging. Even for humans, aging has only become common in recent decades.
At the start of the 20th century, infectious diseases such as pneumonia and influenza caused more deaths in the United States than “organic” diseases like cancer. Now the situation is reversed. The availability of effective weapons against infectious disease (e.g., sanitation, antibiotics, and immunization) has greatly increased the average life span (but not maximum life span) and resulted in “organic” diseases like cardiovascular disease and cancer becoming the most common cause of death.
In 1900, a newborn child in the U.S. could look forward to an average life expectancy of only 47 years. Infectious diseases were the major causes of death, killing most people before they reached an age when aging set in. Three-quarters of a century later, life expectancy had risen to 73 years and “organic” diseases, including all the diseases of aging, had replaced infectious diseases as the major cause of death. Today, the life expectancy has risen to 80 for women (74 for men), and coping with an aging population has become a major economic and social challenge in the U.S.
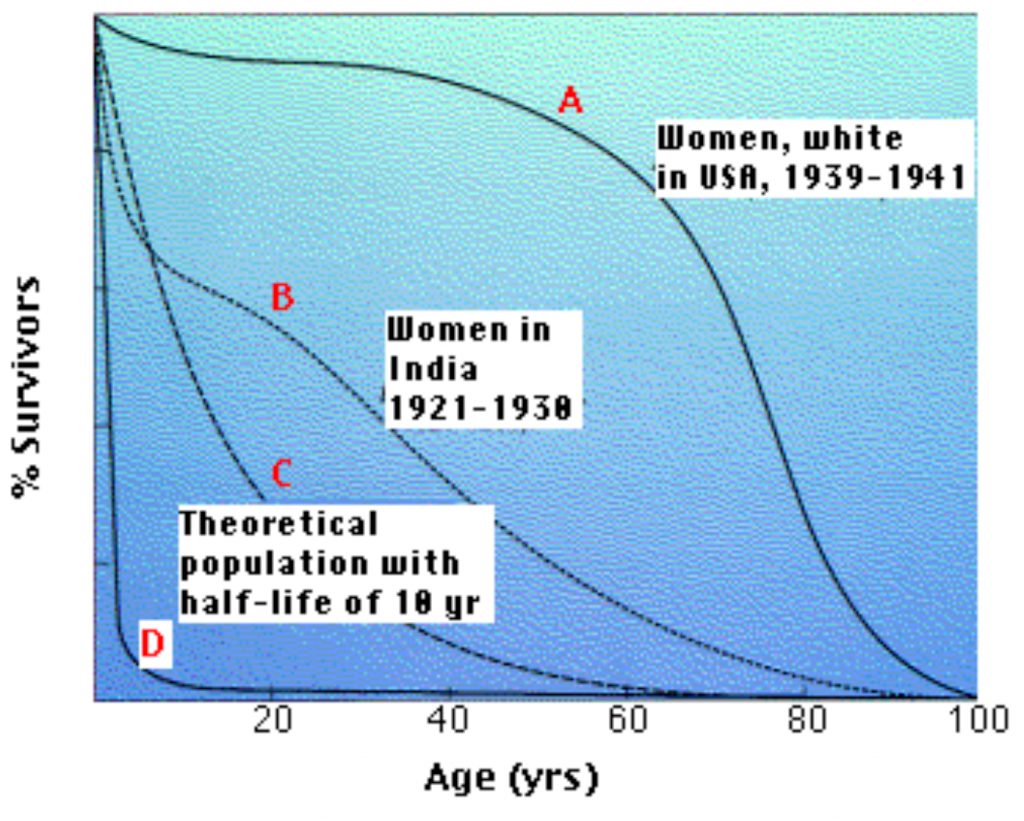
The above graph (Figure 1) shows four representative survival curves. The vertical axis represents the fraction of survivors at each age (on the horizontal axis).
- Curve A is characteristic of organisms that have low mortality until late in life. Then mortality increasingly becomes the endpoint of the aging process.
- Curve B is typical of populations in which such environmental factors as starvation and disease obscure the effects of aging (and infant mortality in high).
- Curve C is a theoretical curve for organisms for which the chance of death is equal at all ages. This might be the case for organisms that show few, if any, signs of aging (some fishes) or those (e.g., songbirds in the wild) that suffer severe random mortality from environmental causes throughout life.
- Curve D is typical of organisms, oysters for example, that produce huge numbers of offspring accompanied by high rates of infant mortality.
Organisms with survivorship curves between C and D have no opportunity to show the signs of aging.
Aging in Invertebrates
Invertebrate animals have provided some important clues about the aging process.
- Colonial invertebrates like sponges and corals don’t show signs of aging. Even individual cnidarians, like the sea anemone that lived for 78 years, show little or no sign of aging. In all these cases, this is probably because there is constant replacement of old cells by new ones as the years go by.
- Lobsters also can live to a very old age with no obvious sign of a decline in fecundity or any other physiological process. But lobsters never stop growing, so once again it may be the continuous formation of new cells that keeps the animal going.
- In culture vessels, Drosophila does have a limited life span and shows signs of aging before it dies. Two factors have been found to influence the aging process and thus life span:
- Calorie restriction, that is, a semi-starvation diet. In fact, restricting food intake has been shown to increase life span (and slow aging) in all animals — including mammals — that have been tested.
- Single genes have been identified that extend life span in Drosophila (and also in the invertebrate Caenorhabditis elegans).
Aging in Vertebrates
Some cold-blooded vertebrates fishes, amphibians, reptiles have long life spans if they can survive environmental hazards (giant tortoises are known to have reached 177 years of age). These animals are cold-blooded and grow so slowly that they will probably succumb to environmental hazards before they stop growing and begin to show signs of aging. The situation is different for birds and mammals. They are warm-blooded, grow rapidly to adult size and, if protected from environmental hazards, will show signs of aging.
Why Do We Age?
Programmed in our genes
The pros
- Single genes have been found that increase life span in Drosophila, C. elegans, and mice. Genes that suppress signaling by insulin and insulin-like growth factor-1 (Igf-1) increase life span in these animals. Examples:
- Mice with one of their Igf-1 receptor genes “knocked out” live 25% longer than normal mice.
- Klotho. Cells in the kidney and brain release the extracellular portion of a cell surface transmembrane protein into the blood. This “hormone”, called Klotho, binds to receptors on many target cells reducing their ability to respond to insulin and Igf-1 signaling.
- Mice homozygous for a mutant klotho gene show many signs of premature aging while
- mice expressing extra-high levels of the Klotho protein live 20–30% longer than normal.
- Long life spans clearly run in human families.
- Aging often appears sooner in animals that suffer high death rates from external causes (e.g. predation) early in life. Why should this be? Three (interrelated) possibilities:
- The accumulation of harmful mutations (in the germline). Few individuals survive long enough for these to be selected against.
- Antagonistic pleiotropy. Genes that promote survival early in life at the expense of maintaining the body will be selected for.
Some examples:- p53. By forcing cells with damaged DNA to stop dividing and become senescent or even to die by apoptosis, it protects the organism from the threat of those cells becoming cancerous but at the expense of reducing cell renewal (e.g., by decreasing the size of the pools of stem cells). Mice that are forced to produce higher-than-normal levels of the p53 protein show many signs of premature aging while female mice that are deficient in p53 have reduced fecundity (blastocysts fail to implant). So here is a tradeoff by a gene that promotes evolutionary success at an early age but at the expense of accelerated aging.
- In both Drosophila and C. elegans, some mutations that increase life span do so at the cost of decreased fecundity and vice versa.
- Disposable soma. Early death from external causes will select for genes that increase the chances of passing germplasm on (i.e. reproduction) at the expense of genes that might delay aging.
- There is no way that natural selection can select for genes whose only beneficial effect appears after the age of reproduction is over. But
- any genes that extend the reproductive period or
- any genes that promote fitness in youth as well as longevity would be selected for.
The cons
High early mortality from external causes (e.g. predators) has been linked to early aging (in the survivors) in some animals, but the reverse has been found in others. These contradictory results do not negate the role of genes in aging, but indicate that other environmental factors (e.g. more food left for the survivors) may skew the outcome.
The Inevitable Consequence of an Active Life
The pros
- Many cold-blooded vertebrates (e.g., many fishes and reptiles) do not show signs of aging.
- Transgenic mice whose “thermostat” in the hypothalamus has been reset to give a lower body temperature (reduced by 0.3 – 0.5°C) live 12% (males) to 20% (females) longer than their nontransgenic littermates. This translates into adding some 3 months to the average life span of 27 months for these mice. (See Conti, B., et al., Science, 3 November 2006).)
- The effects of Calorie Restriction (CR). The life span of yeast, C. elegans, Drosophila, birds, and mammals (mice, rats, and probably monkeys) can be extended, and signs of aging delayed, if they are maintained on a semi-starvation diet. Calorie restriction in mice causes
- a drop in
- the level of circulating insulin and insulin-like growth factor-1 (Igf-1);
- the level of glucose and triglycerides in the blood;
- the level of NADH (produced by cellular respiration) within cells;
- an increase in the production of sirtuins — deacetylases that remove acetyl groups from proteins.
- apoptosis of cells to be inhibited;
- formation of adipose tissue to be suppressed;
- increased production of nitric oxide (NO) which is essential for the benefits of CR to take effect.
- greatly increased physical activity and
- lower body weight.
The life-extending and other effects of CR on rats and mice may not be as significant as they seem. All the studies have been done on laboratory rats and mice. Control animals are normally fed ad libitum, which means that they have food available all the time. But this would not likely be the case in the wild so it may be that the physiology and life span of these animals are already compromised and that the effects of CR are mainly restoring normal conditions for these species.
The problem of proper controls may also explain the divergent results in studies of CR in rhesus monkeys.
- In the 10 July 2009 issue of Science, researchers at the Wisconsin National Primate Research Center reported the status of rhesus monkeys that had been on CR for 20 years compared with a control group allowed to feed ad libitum on the same diet during that time. The results: the CR animals showed markedly fewer signs of aging (none showed any signs of diabetes), and the few that died from age-related causes did so at only one-third the rate of the controls.
- However, a 25-year study of CR in rhesus monkeys carried out at the National Institute of Aging (NIA) (and reported in Nature in August 2012) showed no increased longevity in the CR monkeys.
The difference may have arisen because the control monkeys in Wisconsin were fed ad libitum while the controls at the NIA were fed a fixed amount. Furthermore, the diet used in the Wisconsin study were much higher in sugar than the diet at the NIA, and the NIA diets also included omega-3 fatty acids and other healthy components absent from the Wisconsin diet. So perhaps the positive effect seen in the Wisconsin study resulted from the CR animals simply getting less of an unhealthy diet than the controls.
- a drop in
Resveratrol
Resveratrol, is a small molecule found in red wine which appears to activate sirtuins mimicking the effects of calorie restriction. Mice given daily doses of resveratrol while indulging in a high-fat diet get fat but avoid the degenerative changes and shortened life span that normally accompany a high-fat diet. But before rushing out to buy red wine, realize that the doses of resveratrol given to the mice were far higher than could be supplied by drinking it. Studies on the effect of resveratrol on extending life span in yeast, Drosophila and C. elegans have produced mixed results.
No one knows for certain why calorie restriction delay aging, but some mechanisms might be because it lowers the level of glucose in the blood and thus the speed with which lipids and proteins suffer from glycation. Advanced glycation end products (AGEs) are molecules that have reduced function because of the haphazard addition of sugars to them. For proteins like collagens and elastin, this results is increasing stiffness of the extracellular matrix (ECM) of blood vessels, joints, heart, kidney, etc. Reducing calorie intake reduces female fecundity (at least in C. elegans, Drosophila, rats and mice). The energy that would have been devoted to producing offspring can be devoted instead to tissue repair and maintenance. Calorie restriction raises the level of sirtuins:
- The SIRT1 protein CR in knockout mice lacking SIRT1 gain no increase in life span.
- plays a key role in repairing DNA damage and so helps protect the integrity of the genome which appears to be essential to longevity.
- It also inhibits the nutrient sensor TOR (“target of rapamycin”) which accelerates aging in mice.
- SIRT1 also inhibits p53 activation thus protecting against mitochondrial damage.
- The SIRT3 protein is found in mitochondria where it inhibits the production of free radicals.
The Free Radical Theory of Aging
A major aspect of metabolism is the oxidation of foodstuffs by the mitochondria. Electron transport in the mitochondria generates reactive oxygen species (“ROS“) such as the superoxide anion (O2−), which generates hydrogen peroxide (H2O2). Although cells contain enzymes for detoxifying these reactive substances (e.g., catalase which breaks down H2O2), they eventually and inevitably damage macromolecules in the cell: proteins; lipids; and probably most important of all, DNA.
Damaged proteins and lipids accumulate in the cell, especially nondividing cells like neurons and muscle, producing aggregates of denatured proteins and an “aging pigment” called lipofuscin (a principal component of ear wax). The accumulation of protein aggregates in striated muscles reduces muscle strength. Protein aggregates accumulate more slowly in the cells of animals on a calorie restricted diet — perhaps as a result of more-efficient autophagy. However, it may be damage to DNA that is the crucial factor in the decline in cell function with age.
The DNA of the mitochondria (mtDNA) may be at special risk. ROS are produced as an inevitable byproduct of electron transport in the mitochondria and thus are generated close to the mtDNA. But the products of these genes are essential for electron transport. So perhaps a positive feedback loop is generated: ROS -> mutations in electron transport genes reducing their efficiency -> more ROS production.
Supporting evidence:
- mtDNA accumulates mutations faster than nuclear DNA, and these show the chemical characteristics of damage by ROS.
- Transgenic mice containing the human gene for catalase (but with the targeting signal that would normally send the protein to peroxisomes replaced with that for mitochondria) live 20% longer than normal for their strain. [See Schriner, S. E. et al., Science, 24 June 2005.]
- Transgenic mice whose DNA polymerase for copying mtDNA genes (DNA polymerase gamma) is defective, and introduces an elevated number of mutations in mtDNA, show many signs of premature aging — both cellular and in various organ systems — and die early.
The cons
- Neither mice genetically engineered to overproduce free radicals, nor those engineered to produce lower amounts of free radicals, have any change in their lifespan.
- Bats and mice are similar in size and metabolic rate, but bats can live ten times as long.
- Although glucose-starved yeast do live longer, they have an increased — not decreased — rate of cellular respiration.
- The metabolic rate of mice on a CR diet is no lower than that of mice on a normal diet.
- The beneficial effects of CR take hold at any time, at least in Drosophila. Even after three weeks on a rich diet (in the second half of the normal life span of adult flies), switching to a CR diet reduces mortality to the same degree as flies maintained on CR throughout their adult lives. The reverse is also true — switching from a CR diet to a rich diet quickly undoes the good work of the former. These results suggest that if a rich diet does produce irreversible and accumulating damage, its harmful effects on life span can be blunted at any time.
The Accumulation of Senescent Cells
Chronological Senescence
Once formed, some cells in a mouse or human are never replaced. A neuron formed during embryonic development may still be functioning at the end of life. However, during its life span, damage to its organelles and DNA may accumulate resulting in a loss of function. This is called chronological senescence. In other tissues, e.g., blood and epithelia, new cells replace old ones throughout life. But even though new, they may have reduced function because of replicative senescence.
Replicative Senescence
One might expect that cells removed from a mouse or human and placed in tissue culture could be cultured indefinitely, but that is not the case. When human fibroblasts, for example, are placed in culture, they proliferate at first, but eventually a time comes when their rate of mitosis slows and finally stops. The cells continue to live for a while, but cannot pass from G1 to the S phase of the cell cycle. This phenomenon is called replicative senescence. Fibroblasts taken from a young human pass through some 60–80 doublings before they reach replicative senescence.
Why should this be? Cells — unless they retain the enzyme telomerase — lose DNA from the tips of their chromosomes (telomeres) with each cell division. In general, the telomeres in the cells of old animals are much shorter than those in young animals. A recent study of short-lived versus long-lived birds showed that telomere shortening was faster in the short-lived species. And one species, a petrel which lives four times as long as other birds of its size, actually has telomeres that grew longer with age. Most somatic cells of the body cease to express telomerase. However, cells genetically manipulated to express telomerase long after they should have stopped, avoid replicative senescence. Germline cells, e.g., spermatogonia, and some stem cells continue to express the enzyme. Some 95% of cancer cells express telomerase. If telomeres get too short (less than 13 repeats in human cells), chromosome abnormalities — a hallmark of cancer — occur. Cancer can be avoided if the cell senses this dangerous condition and ceases to divide. So telomere shortening may protect against cancer at the price of cell senescence.
Two proteins encoded by tumor suppressor genes p53 and p16INK4a play pivotal roles in stopping the cell cycle. The result: replicative senescence. So replicative senescence may be the price we pay for removing cells from the cell cycle before they can accumulate the mutations that would turn them into cancer cells.
The role of the tumor suppressor proteins in replicative senescence is mirrored in the intact animal, at least in mice.
- Mice engineered to express abnormally-high levels of p53 activity show many signs of premature aging including premature reduction in the length of their telomeres.
- Mice that express abnormally-high levels of p16INK4a have a reduced ability to regenerate tissue while mice whose activity of p16INK4a is suppressed continue to repair damaged tissue as efficiently as young animals do.
- In mice, eliminating senescent cells (they are high in p16INK4a) prevents (in young mice) and partially reverses (in older mice) some of the signs of aging such as cataracts, and loss of adipose tissue and skeletal muscle mass.
- Mice genetically-engineered to express high levels of telomerase and tumor suppressor genes (e.g., p53 and p16INK4a) have substantially-increased life spans and show fewer of the degenerative changes characteristic of aging in their skin and other epithelia. (The need to make these transgenic mice with increased tumor suppressor activity in addition to increased telomerase activity arises because an increased level of telomerase alone elevates the incidence of cancer — killing the animals before any anti-aging effects of telomerase can be measured reliably.)
The role of telomerase deficiency in mammalian aging
Mice whose genes for telomerase have been “knocked out” (either Tert−/− or Terc −/−) show many of the degenerative changes associated with aging.
- The number of mitochondria in their cells decreases as does the function of those that remain.
- Oxygen consumption and ATP production declines.
- The efficiency of the electron transport chain decreases.
- This leads to an increased generation of reactive oxygen species (ROS).
- The level of p53 activity increases.
- mitosis declines
- apoptosis of cells increases
- replicative senescence increases
- The anatomy and function of organs such as the liver and heart show the degenerative changes of age.
In the 6 January 2011 issue of Nature, Mariela Jaskelioff and her colleagues (many of the same team that found the results described in the previous section) report that reactivation of telomerase in aged mice reverses many signs of aging.
Their experimental animals were another telomerase-deficient strain of mice; that is, mice that couldn’t produce telomerase even in those cells — “adult” stem cells and cells of the germline — that normally retain telomerase activity. The mice were made by “knocking-in” a gene that prevents any expression of telomere reverse transcriptase (TERT) unless an activating drug is given to the animal. Without the drug, these mice live half as long as normal, and as they get older, they display many signs of aging:
- their telomeres get shorter leading to chromosome aberrations;
- their cells undergo early replicative senescence;
- almost all their organs — testes, spleen, intestine, brain — show degenerative changes typical of aging.
BUT, if given the activating drug over a four-week period at a time when degenerative changes were already apparent (25-30 weeks), their deterioration stopped and even partially reversed.
- the length of their telomeres increased;
- replicative senescence was delayed;
- their life span was substantially increased;
- their brain, testes, liver, spleen, and intestine escaped the degenerative changes seen in untreated telomerase-deficient mice;
- they produced larger litters than untreated mice;
- there was reduced activation of p53, indicating
- reduced damage to their genome and
- reduced apoptosis in their tissues
How would replicative senescence of cells lead to the deterioration in structure and function of the aging tissues (e.g., skin) in which they reside? In tissues, e.g., skin and other epithelia, where mitosis must continue throughout life to replace the cells that are lost, the accumulation of senescent cells — incapable of further mitosis — could leading to the characteristic changes of aging in that tissue.
- One mechanism could be simply the inability of senescent cells to repair the tissue by mitosis.
- However, senescent cells remain active although the genes they express change. Perhaps the proteins they secrete (e.g., collagen-digesting enzymes) cause the aging changes in the tissue where they reside.
- Perhaps it is the senescence of adult stem cells that has the greatest effect on tissue aging. In knockout mice that cannot make the Klotho protein, stem cells and progenitor cells in various tissues undergo senescence and decline in numbers.
An unavoidable tradeoff?
Some of the data so far suggests that efforts to avoid the degenerative changes that come with age (e.g., by increasing cell renewal by means of increased telomerase activity) in the hope of increasing longevity may instead hasten death from cancer while efforts to prevent cancer (e.g., by increasing the activity of tumor suppressor genes) may hasten aging. However, other evidence paints a less-gloomy picture. Mice heterozygous for the p53 tumor suppressor gene (p53+/−) develop many cancers when exposed to ionizing radiation. With only a single copy of this tumor suppressor, a single cell is at great risk of losing the remaining copy (“loss of heterozygosity”) and starting the growth of a malignant clone. However, if before being irradiated the mice are given resveratrol — to stimulate the production of the anti-aging SIRT1 protein — the incidence of some cancers is reduced and the mice live longer before succumbing to their tumors.
The Accumulation of Genetic Errors
The pros
- Mice given ionizing radiation that damages DNA show early aging.
- Transgenic mice with a defect in the “proofreading” function of the DNA polymerase responsible for copying mitochondrial DNA accumulate many mutations in their mitochondrial genes and show marked signs of premature aging.
- Cells taken from old mice (and old humans) show slightly elevated levels of somatic mutations and chromosome abnormalities like translocations and aneuploidy. Many of these changes also cause cancer so it is no accident that the incidence of cancer rises with advancing age (graph).
- The hematopoietic stem cells of “knockout” mice deficient in any one of these enzymes needed for genome maintenance
- XPD for nucleotide excision repair (NER)
- Ku80 for nonhomologous end joining (NHEJ)
- TR (telomerase RNA) needed for telomere maintenance
lose their ability to supply the various progenitor cells that produce the white blood cells.
- Most of the hematopoietic stem cells in aged mice show evidence of double-stranded breaks (DSBs) in their chromatin.
- As DSBs form, SIRT1 proteins move from their original locations (at gene promoters) to the locations of the DSBs (where they recruit DNA repair proteins). This shifts the pattern of gene expression to one typical of aging cells.
- Cells taken from old people (and people with premature aging syndromes) show marked reductions in the transcription of some genes, increases in others.
Clues from the Transcriptome of Aging Brains
A group of Harvard researchers reported (in the 26 June 2004 issue of Nature) the results of their study of gene expression in the human brain. They extracted the RNA from autopsied brain tissue of 30 people who had died at ages ranging from 26 to 106. They analyzed the RNA with DNA chips looking for the level of activity of some 11,000 different genes (the transcriptome). A clear pattern emerged.
The level of activity of some 400 genes changed over time.
- Gene expression declined in old age for many genes. Some examples:
- genes encoding proteins involved in synaptic activity in the brain (e.g., learning, memory)
- NMDA, AMPA, GABAA receptors
- calcium-calmodulin-dependent kinase II (CaMKII)
- genes involved in mitochondrial functions, such as
- production of ATP (needed for DNA repair)
- production of damaging reactive oxygen species (ROS)
- genes encoding proteins involved in synaptic activity in the brain (e.g., learning, memory)
Detailed examination of some of these down-regulated genes showed that they had suffered DNA damage — more often in their promoters than in their coding regions.
- Gene expression increased in old age for other genes. Some examples:
- genes involved in inflammation and other immune defenses;
- genes encoding proteins involved in defense against reactive oxygen species (ROS);
- genes encoding proteins involved in DNA repair.
The transition from the youthful transcriptome to the transcriptome of the aged brain occurred at varying times from as young as 42 to as old at 73.
A study of individual heart muscle cells in young and old mice (Bahar, R. et al., Nature, 22 June 2006) showed that the transcriptome of young cells was quite uniform from cell to cell but that of aged cells was highly variable from one cell to another. Variable gene expression from one cell to the next in a single tissue might well lead to defects in the functioning of that tissue.
Clues from Premature Aging Syndromes
Humans suffer from a number of rare genetic diseases that, among other things, produce signs of premature aging, e.g., gray hair, wrinkled skin, and shortened life span. In several cases, the mutated genes are ones that have roles to play in maintaining the integrity of the genome, that is, in DNA repair.
- Werner’s syndrome. The hair of patients turns gray in their 20s and most die in their late 40s with such signs of age as osteoporosis, cataracts, and atherosclerosis. Even when young, their cells undergo replicative senescence after only ~20 doublings instead of the normal 70 or more. Caused by mutations in WRN, which encodes a helicase needed for DNA repair and maintenance of telomeres.
- Cockayne syndrome (CS). Caused by mutations in genes needed for DNA repair, especially transcription-coupled DNA repair. While these people show only some of the signs of aging, they do have a sharply-reduced life span.
- Ataxia telangiectasia (AT). These patients show signs of premature aging. They lack a functioning gene (ATM) product needed to detect DNA damage and initiate a repair response.
- Hutchinson-Gilford progeria syndrome. Children with this rare disorder show many signs of severe aging by their second birthday and die in their early teens. Caused by mutations in the gene (LMNA) for lamin the intermediate filament protein that stabilizes the inner membrane of the nuclear envelope. The machinery for DNA replication, transcription, and repair is located at the inner surface of the nuclear envelope, and the cells of these patients have increased DNA damage and other defects in gene expression.
So these syndromes suggest that aging may be the consequence not so much of mutations in general, but of mutations in those genes whose products are essential for the error-free replication, repair and transcription of all genes.
Why is a mouse as old at 2 years as a human at 70
If aging represents the inevitable consequence of a failure of DNA repair, why does it occur so much sooner in some mammals (e.g., mice) than in others (e.g., elephants and humans)?
The answer probably lies in the risk of death from external factors (e.g., predation, starvation, cold) in that species.
As noted above, few small mammals ever age because they die early of external causes. These animals are r-strategists, putting their energy into quickly
- reaching sexual maturity
- producing large numbers of offspring that can soon live independently
There is no selective advantage for them to invest in the machinery of efficient DNA repair because they are going to die before mutations become a problem.
Humans, in contrast, are K-strategists. They take a long time to reach sexual maturity. They also produce small numbers of young that must be cared for over a long period. Small wonder, then, that evolution in humans (and other long-lived mammals) has selected for genes promoting efficient DNA repair.
Table 2 shows that the efficiency of DNA repair is directly correlated with life span in a variety of mammals.
| Species | Average life span, yr | Relative effectiveness of DNA repair |
| Human | 70 | 50 |
| Elephant | 60 | 47 |
| Cow | 30 | 43 |
| Hamster | 4 | 26 |
| Rat | 3 | 13 |
| Mouse | 2 | 9 |
| Shrew | 1 | 8 |
Interrelationships
Examining the various factors that have been implicated in the aging process suggests that most —perhaps all — are interrelated.
- Mitochondria disfunction with the production of…
- reactive oxygen species (ROS) with their damaging effect on…
- DNA and other cell constituents coupled with the…
- onset of replicative senescence so that damaged cells can no longer be replaced.
All of the above may all play important roles. So the factors described above are by no means mutually exclusive.
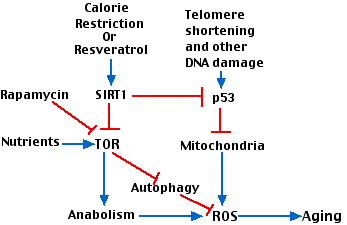
Figure 2 attempts to show how various factors involved in aging interact. Key players are:
- The gradual shortening of telomeres with repeated cell divisions
- p53
- the enzyme designated TOR (“target of rapamycin”). TOR is a kinase that participates in many metabolic pathways in the cell. (It is inhibited by the antibiotic rapamycin that is used as an immunosuppressant)
Stimulatory interactions are shown with blue arrows; inhibitory interactions are shown in red. Interactions include:
- Telomere shortening activates p53 which leads to damaged mitochondria.
- The inefficient electron transport chain in damaged mitochondria produces ROS.
- Abundant nutrients (e.g. amino acids) as well as other growth stimulants activate TOR which promotes anabolism (protein and lipid synthesis) with attendant production of reactive oxygen species (ROS) and aging.
- Calorie restriction, working through SIRT1 inhibits TOR and its downstream effects.
- Inhibition of TOR relieves its inhibition of autophagy allowing the cells to scavenge, for example, damaged mitochondria.
- SIRT1 inhibits p53 activation thus protecting against mitochondrial damage.
Because of the association between telomere shortening and aging, two companies have begun (in 2011) to offer tests of telomere length. How such tests might be useful to the people asking for them remains to be seen.
The Hallmarks of Aging
In the 6 June 2013 issue of Cell, an international group of scientists developed a list of 9 features that characterize aging in animals. (This effort to bring order to such a complex subject is reminiscent of earlier papers in the same journal, The Hallmarks of Cancer.)
They expected that each hallmark would meet at least two of three criteria.
- It should be characteristic of normal aging.
- Increased expression of the hallmark should result in faster aging.
- Efforts to reduce expression of the hallmark should prolong a healthy lifespan (“healthspan”).
The 9 hallmarks of aging
1. Genomic Instability
Meeting the criteria:
- Aged cells contain more DNA damage than young ones.
- Agents that increase unrepaired damage to DNA, including chromosomal damage (e.g., aneuploidy), hasten aging.
- Limited evidence that treatments that reduce, for example, chromosome missegregation, prolong healthspan.
2. Telomere Attrition
Meeting the criteria:
- The chromosomes of aged cells have shorter telomeres than those of young cells.
- Telomerase-deficient mice show premature aging.
- Treatments that reactivate telomerase in normal mice delay aging.
3. Epigenetic Alterations
Meeting the criteria:
- The patterns of DNA methylation and histone modifications changes as a mammal ages.
- Mice that are deficient in the sirtuin SIRT6, an enzyme that deacetylates histones, age more rapidly than normal.
- Treatments that increase the activity of sirtuins increase healthspan in mice.
Note
As we humans age, the DNA in our cells accumulates an ever-increasing number of epigenetic changes as measured by the methylation of CpGs. This is true for a wide variety of cell types even those that have been formed recently; that is, the number of epigenetic changes reflects the age of the donor not the age of the cell. The correlation is so good that analysis of these changes in a cell can predict the donor’s age sometimes within a matter of months.
4. Loss of Proteostasis
Proteostasis is the homeostasis of the proteome — the proper balance of the synthesis and degradation of proteins in the cell.
Meeting the criteria:
- The clearance of denatured (unfolded) proteins by autophagy and proteasomes, as well as the ability to refolded them with chaperones, all decline with age. The result: toxic protein aggregates that accumulate in aged cells.
- Mutant mice with defective chaperone activity age more quickly.
- Transgenic Drosophila and C. elegans that overexpress chaperones have increased life spans.
5. Derugulated Nutrient Sensing
Meeting the criteria:
- The nutrient sensor TOR (“target of rapamycin”), which promotes anabolism, increases during normal aging (and produces obesity, at least in mice).
- Increased activity of TOR accelerates aging in mice.
- Examples:
- Genetic suppression of TOR signaling extends lifespan in Drosophila and C. elegans.
- Calorie Restriction (CR), which inhibits TOR, increases healthspan in all animals in which it has been tested.
- Rapamycin, which inhibits TOR, extends lifespan in Drosophila, C. elegans, and mice.
6. Mitochondrial Dysfunction
Meeting the criteria:
- The production and efficiency of mitochondria decreases in aging, otherwise normal, mice.
- Deleterious mutations in mitochondrial DNA and other defects in mitochondrial function all accelerate aging in mice.
- No compelling evidence yet that treatments to improve mitochondrial function increase life span.
7. Cellular Senescence
Meeting the criteria:
- Replicative senescence (cells no longer able to enter the cell cycle) sets in much sooner in the cells of aged animals than it does in young animals.
- Mice engineered to express abnormally-high levels of p53, a protein that blocks entry into the cell cycle, show many signs of premature aging.
- In mice, eliminating senescent cells prevents (in young mice) and partially reverses (in older mice) some of the signs of aging such as cataracts, and loss of adipose tissue and skeletal muscle mass.
8. Stem Cell Exhaustion
Meeting the criteria:
- The proliferative capacity of adult stem cells declines with age in the tissues that have been examined.
- Deliberately exhausting the pool of stem cells in the Drosophila intestine leads to premature aging.
- Transplantation of stem cells from young mice into aged mice improves the degenerative changes of aging and prolongs their life.
9. Altered Intercellular Communication
All cells respond to chemical signals in their environment. These include cytokines secreted by nearby cells (paracrine stimulation).
Meeting the criteria:
- Inflammation in various tissues — mediated by the secretion of a variety of cytokines — increases in the aged.
- Genetically engineered mice that are unable to down-regulate the mRNAs synthesizing pro-inflammatory cytokines show accelerated aging.
- Inhibition of the pro-inflammatory cytokine NF-κB delays aging in mice. Even such a simple anti-inflammatory agent as aspirin seems to prolong life in mice.
Relationships of the Hallmarks
- The first 4 hallmarks appear to represent the initiating events leading to aging.
- Hallmarks 5, 6, and 7 appear to represent damage produced as the cell attempts to respond to the damage caused by the first 4 hallmarks.
- Taken together, hallmarks 1 through 7 produce the aging phenotype seen in hallmarks 8 and 9, which are ultimately responsible for the decline with age in the function of cells and the organism of which they are a part.
An Elixir of Youth?
Despite years of research, only three interventions have been discovered that slow the aging process and/or prolong life.
- Calorie Restriction (CR). Works in all animals tested.
- Rapamycin. Extends lifespan in mice, Drosophila, and C. elegans but does not appear to reverse or even halt the degenerative changes of aging.
- Parabiosis. When the circulatory system of a young mouse is joined to that of an old mouse (the technique is called parabiosis), various tissues in the old mouse, e.g., its skeletal muscle, cardiac muscle, liver, and central nervous system, become rejuvenated. A promising candidate for mediating this effect is a protein called GDF11 (“Growth Differentiation Factor 11”). (GDF11 is also known as BMP-11). Injections of recombinant GDF11 are almost as effective as parabiosis. GDF11 probably acts by stimulating the activity of stem cells. However, there is no evidence yet that it increases the lifespan of the mice.
Aging in Unicellular Organisms
It used to be thought that many unicellular organisms, such as yeast and bacteria, were immortal; that is, they
- never aged but simply kept dividing to produce new individuals;
- did not have the distinction between germline (immortal) and soma (mortal) that multicellular organisms have.
If true, every time a cell divides, the two daughter cells would be identical in all respects to the parent (symmetrical division).
But at least for yeast and E. coli, this is not the case.
Yeast cells do age and, as discussed above, have proved useful for studying the aging process. Placing a single yeast cell on solid medium and removing its daughter (the bud) each time one is produced, it turns out that the number of times the mother cell can form a new bud by mitosis is limited. After producing a bud some 20–30 times, the mother cell shows a number of harmful cellular changes (e.g., defective mitochondria) and dies.
But, at least early in her life, the buds are born with the potential of a full life span. So the mitotic division must be asymmetrical with the properties of the bud different from those of its parent. Several mechanisms by which this occurs have now been demonstrated.
- A diffusion barrier is formed at the neck between the mother cell and her bud. This barrier prevents the passage from the mother into the bud of damaged nuclear components, e.g., plasmid-like fragments of DNA produced during the life of the mother. In contrast to the situation in most eukaryotes, in yeast there is no breakdown of the nuclear envelope during mitosis. During anaphase, the nuclear envelope grows and the portion enclosing the daughter chromosomes enters the bud. However, the barrier at the neck keeps all the preexisting nuclear pore complexes (NPCs) within the mother cell and these retain the DNA fragments and perhaps other damaged nuclear components.
- Most of any damaged cytoplasmic components, such as proteins denatured by reactive oxygen species (ROS) resulting in the formation of nonfunctional aggregates, become attached to aged mitochondria and the endoplasmic reticulum both of which are retained in the mother cell. Any protein aggregates that do get through are either dissolved by chaperones in the bud or, if that fails, actin filaments in the bud move the aggregates back into the mother cell. This latter mechanism requires the presence of a number of proteins, including a sirtuin found in yeast called Sir2 (“Silent information regulator 2”).
Asymmetric division in stem cells
Stem cells are cells that divide asymmetrically to produce a daughter cell that goes on to differentiate and a daughter cell that remains a stem cell. A number of examples have been found — in Drosophila and in mammals — where aging stem cells preferentially deposit their damaged cellular components, e.g. aggregated proteins, in the daughter that will go on to differentiate while keeping undamaged components in the daughter that will remain a stem cell. So like yeast, these stem cells have a mechanism that preferentially protects the “immortal” cell from the inevitable effects of aging.
Aging in E. coli
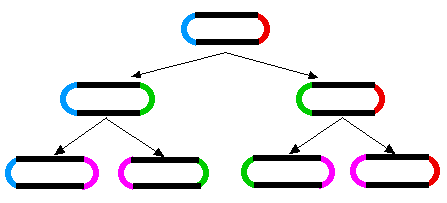
A similar aging phenomenon has been found in E. coli. When E. coli divides, a septum forms in the middle of the dividing cell and then the two daughter cells are pinched apart. As the cell wall seals the break, the two daughter cells end up with one “old” end and one newly-formed end. When the two daughters go on to divide, the process is repeated. The original old ends gets passed on from generation to generation (rather like immortal strands of DNA).
The diagram (Figure 3) shows, how during cell division, two new poles are formed, one in each of the progeny cells (new poles shown in green for the first generation; magenta for the second). The other ends of those cells were formed during a previous division.
It has been shown (Stewart EJ, Madden R, Paul G, Taddei F (2005) Aging and Death in an Organism That Reproduces by Morphologically Symmetric Division. PLoS Biol 3(2): e45 doi:10.1371/journal.pbio.0030045) that the cells that inherit an increasingly old pole exhibit a diminished growth rate, decreased offspring production, and an increased incidence of death.
So it appears that the phenomenon — characteristic of all multicellular organisms — of an aging and mortal soma producing germplasm (sperm and eggs) that starts a new youthful generation may have its counterpart in unicellular organisms. Perhaps no single cell can escape the ravages of time on the integrity of its organelles and the molecules.
Aging in Plants
Annuals and Biennials
Annuals, such as many grasses and “weeds” grow vigorously for a period, then form flowers, followed by fruits. Fruiting is followed by a slowing of growth accompanied by physiological and morphological changes such as:
- an increase in the rate of respiration (catabolism)
- loss of chlorophyll
These changes constitute aging and end in the death of the plant. Biennials follow the same pattern, but take two years to do it. This pattern in clearly programmed in the genes. Even with plentiful moisture, soil minerals, sunlight, and warm temperatures, the plants age and die.
Perennials
The situation is quite different in perennials. Throughout their lives, woody perennials (trees) produce new vascular tissue, leaves, and flowers each year. They do not show marked signs of aging, although their rate of growth may decline over the years. Finally, disease or inability to support their ever-increasing size against wind or snow load lead to their death.

Figure 4 (courtesy of Walter Gierasch) shows a bristlecone pine (Pinus longaeva) growing in the White Mountains of eastern California. Tree-ring analysis shows that some of these trees are almost 5000 years old. But note that no living cells in the tree are more than a few years old.
Even so, how have long-lived plants like these avoided the accumulation — over years of DNA replication as their cells divided — of deleterious mutations that would reduce fitness and life span? Perhaps it is because the cells in plant meristems, where all growth begins are stem cells which divide slowly and like all stem cells, asymmetrically; that is, producing one daughter that will remain a stem cell and one that will begin a phase of rapid mitosis and eventually differentiate into the mature tissues of the plant. If (and this is as yet only a speculation) the division of a meristematic stem cell is asymmetric with respect to the segregation of DNA strands; that is, the stem cell retains the immortal strands of DNA while the cell destined to produce more tissues receives the newly-replicated strands, this would provide an additional mechanism to protect the genome as the years go by.
This chapter by John W. Kimball is licensed CC BY 3.0.
